Il modello autosomico recessivo caratterizza le malattie causate da mutazioni
a carattere recessivo, sono perciò necessarie due copie dell’allele mutato perché si manifesti la malattia.
In questo tipo di trasmissione la figura chiave è quella del portatore sano.
Il portatore sano è un soggetto eterozigote per la mutazione
, non si ammala ma la trasmette alla discendenza.
La caratteristica epidemiologica di queste malattie è la comparsa di individui malati che hanno gli ascendenti sani.
Gli alberi genealogici delle malattie autosomiche recessive hanno le seguenti caratteristiche:
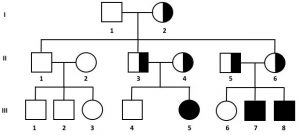
1. spesso la malattia compare in un albero genealogico privo di casi di malattia, cioè individui affetti nascono da portatori sani;
2. i portatori sani possono essere sia maschi che femmine;
3. I maschi e le femmine possono essere affetti con uguale probabilità;
4. vi possono essere dei salti generazionali della malattia e per questo il pedigree viene definito orizzontale.
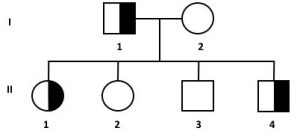
5. dall’accoppiamento tra un portatore sano e un individuo non portatore della mutazione
si avrà una prole con il 50% di portatori sani;
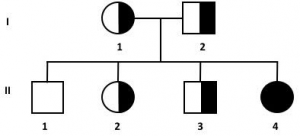
6. dall’accoppiamento tra 2 portatori sani si avrà una prole costituita dal 25% di individui omozigoti recessivi malati, dal 50% di individui eterozigoti portatori sani e dal 25% di individui sani non portatori della mutazione
;
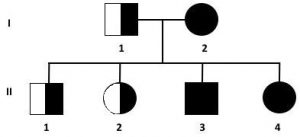
7. L’accoppiamento tra un portatore sano, eterozigote, e un individuo malato, omozigote, determina la comparsa della malattia nel 50% dei discendenti e la condizione di portatore sano, eterozigote, nel restante 50% dei discendenti.
8. dall’accoppiamento di due individui malati il 100% dei figli sarà malato, come per il modello autosomico dominante
quando una dei genitori è omozigote, ma come visto questo genotipo è raro e spesso determina morte embrionale.
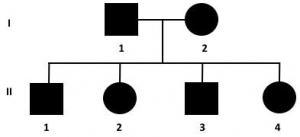
Gli ultimi 2 accoppiamenti sono rari ma non impossibili quando la malattia, come per le autosomiche dominanti, ha un periodo di latenza clinica compatibile con la riproduzione in perfetta salute degli animali.
9. L’accoppiamento tra individui parenti aumenta la probabilità di incontro tra individui eterozigoti.
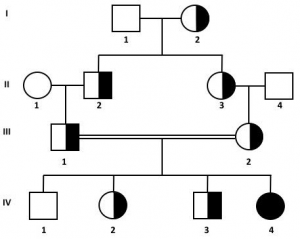
Nell’analisi della genealogia, per stabilire se si tratta di una malattia autosomica recessiva, può essere importante verificare se c’è parentela tra gli individui che hanno dato origine agli animali malati.
La diagnosi di malattia autosomica recessiva richiede una attenta valutazione del pedigree con l’individuazione di uno dei modelli sopra indicati.
Con i test genetici è possibile individuare i portatori sani della malattia e la condizione di latenza clinica della malattia che può manifestarsi nelle anche malattie autosomiche recessive anche se con una frequenza minore rispetto a quelle autosomiche dominanti.
- Acidemia L-2-Idrossiglutarica / L-2-hydroxyglutaricacidemia: Gene L2HGDH (OMIA 001371-9615)
- Acrodermatite letale / Acrodermatitis, lethal: Gene MKLN1 (OMIA 002146-9615)
- Alfa fucosidosi / Fucosidosis, alpha: Gene FUCA1 (OMIA 000396-9615)
- Amelogenesi imperfetta / Amelogenesis imperfecta: Gene ENAM (OMIA 001805-9615)
- Amelogenesi imperfetta | Amelogenesis imperfecta: Gene ACP4 (OMIA 002177-9615)
- Amelogenesi imperfetta | Amelogenesis imperfecta: Gene SCL24A4 (Volume 4 of Canine Medicine and Genetics)
- Caduta episodica / Episodic falling: Gene BCAN (OMIA 001592-9615)
- Cataratta ad insorgenza precoce / Cataract, early onset: Gene HSF4 (OMIA 001758-9615) 1
- Cheratocongiuntivite secca congenita e dermatosi ittiosiforme / Congenital keratoconjunctivitis sicca and ichthysiformis dermatosis: Gene FAM83H (OMIA 001683-9615)
- Cistinuria di tipo I - A / Cystinuria, type I - A: Gene SLC3A1 (OMIA 000256-9615)
- Collasso indotto dall'esercizio fisico / Exercise Induced Collapse: Gene DNM1 (OMIA 001466-9615)
- Deficienza del fattore VII / Factor VII deficiency: Gene F7 (OMIA 000361-9615)
- Deficienza del fattore XI / Factor XI deficiency: Gene F11 (OMIA 000363-9615)
- Deficienza della piruvato chinasi degli eritrociti / Pyruvate kinase deficiency of erythrocyte: Gene PKLR (OMIA 000844-9615)
- Deficienza della piruvato deidrogenasi / Pyruvate dehydrogenase deficiency: Gene PDP1 (OMIA 001406-9615)
- Deficienza di adesione dei leucociti di tipo I / Leukocyte adhesion deficiency, type I: Gene ITGB2 (OMIA 000595-9615)
- Deficienza di adesione dei leucociti di tipo III / Leukocyte adhesion deficiency, type III: Gene FERMT3 (OMIA 001525-9615)
- Deficienza di C3 / C3 deficiency: Gene C3 (OMIA 000155-9615)
- Discinesia ciliare primaria / Ciliary dyskinesia, primary: Gene CCDC39 (OMIA 001540-9615)
- Discinesia parossistica / Dyskinesia, paroxysmal: Gene PIGN (OMIA 002084-9615)
- Displasia ectodermica/sindrome da fragilità cutanea / Ectodermal dysplasia/skin fragility syndrome: Gene PKP1 (OMIA 001864-9615)
- Distrofia corneale maculare / Macular corneal dystrophy: Gene LOC489707 (OMIA 002071-9615)
- Distrofia muscolare tipo la distrofia muscolare di Ullrich nell'uomo / Muscular dystrophy, Ullrich type: Gene COL6A1 (OMIA 001967-9615)
- Epidermolisi bollosa distrofica / Epidermolysis bullosa, dystrophic: Gene: COL7A1 (OMIA 000341-9615)
- Epidermolisi bollosa giunzionale / Epidermolysis bullosa, junctionalis: Gene LAMA3 (OMIA 001677-9615)
- Epidermolisi bollosa semplice / Epidermolysis bullosa, simplex: Gene PLEC (OMIA 002080-9615)
- Gangliosidosi GM1 / Gangliosidosis GM1: Gene GLB1 (OMIA 000402-9615)
- Gangliosidosi GM2 di tipo 1 (variante B) / Gangliosidosis GM2, type I (B variant): Gene HEXA (OMIA 001461-9615)
- Gangliosidosi GM2, di tipo II (variante di Sandhoff o variante 0) / Gangliosidosis GM2, type II (Sandhoff or variant 0)
- Iperossaluria primaria di tipo I (Ossalosi I) / Hyperoxaluria, primary, type I (Oxalosis I): Gene AGXT (OMIA 001672-9615)
- Ipofosfatasia / Hypophosphatasia: Gene ALPL (OMIA 002162-9615)
- Ipomineralizzazione dentale / Dental hypomineralization: Gene FAM20C (OMIA 002015-9615)
- Ipotiroidismo / Hypothyroidism: Gene TPO (OMIA 000536-9615)
- Leucoencefalomielopatia / Leukoencephalomyelopathy: Gene NAPEPLD (OMIA 001788-9615)
- Malassorbimento intestinale della cobalamina / Intestinal cobalamin malabsorption: Gene AMN (OMIA 000565-9615)
- Malassorbimento intestinale della cobalamina / Intestinal cobalamin malabsorption: Gene CUBN (OMIA 001786-9615)
- Malattia da accumulo di glicogeno Ia / Glycogen storage disease Ia: Gene G6PC (OMIA 000418-9615)
- Malattia da accumulo di glicogeno II / Glycogen storage disease II: Gene GAA (OMIA 000419-9615)
- Malattia da accumulo di glicogeno IIIa / Glycogen storage disease IIIa: Gene AGL (OMIA 001577-9615)
- Malattia da accumulo di glicogeno VII / Glycogen storage disease VII: Gene PFKM (OMIA 000421-9615)
- Malattia di Krabbe / Krabbe disease: Gene GALC (OMIA 000578-9615)
- Malattia di Von Willebrand di tipo II / Von Willebrand disease II: Gene VWF (OMIA 001339-9615)
- Malattia di Von Willebrand di tipo III / Von Willebrand disease III: Gene VWF (OMIA 001058-9615)
- Malattia di Wilson / Wilson disease: Gene ATP7B (OMIA 001071-9615)
- Malattia di Wilson correlata al gene COMMD1 / Wilson disease, COMMD1 type: Gene COMMD1 (OMIA 001988-9615)
- Membranite lignea / Ligneous membranitis: Gene PLG (OMIA 002020-9615)
- Metaemoglobinemia / Methaemoglobinaemia: Gene CYB5R3 (OMIA 002131-9615)
- Mielopatia degenerativa / Degenerative myelopathy: Geni SOD1, SP110 (OMIA 000263-9615)
- Miotonia / Myotonia: Gene CLCN1 (OMIA 000698-9615)
- Narcolessia / Narcolepsy: Gene: HCRTR2 (OMIA 000703-9615)
- Neuropatia sensoriale / Neuropathy, sensory: Gene FAM134B (OMIA 002032-9615)
- Neutropenia ciclica / Neutropenia, cyclic: Gene AP3B1 (OMIA 000248-9615)
- Sindrome da mutilazione acrale / Acral mutilation syndrome: Gene GDNF (OMIA 001514-9615)
- Sindrome di Bernard-Soulier di tipo C / Bernard-Soulier syndrome, type C: Gene GP9 (OMIA 002207-9615)
- Sindrome di Musladin-Lueke / Musladin-Lueke syndrome: Gene ADAMTSL2 (OMIA 001509-9615)
- Sindrome di Scott / Scott Syndrome: Gene ANO6 (OMIA 001353-9615)
- Sindrome miastenica congenita / Myasthenic syndrome, congenital: Gene CHAT (OMIA 002072-9615)
- Sindrome miastenica congenita / Myasthenic syndrome, congenital: Gene CHRNE (OMIA 000685-9615)
- Sindrome miastenica congenita / Myasthenic syndrome, congenital: Gene COLQ (OMIA 001928-9615)
- Tromboastenia / Thrombasthenia: Gene ITGA2B (OMIA 001000-9615)
- Trombopatia | Thrombopathia: Gene RASGRP1 (OMIA 001003-9615)